انواع رنگ آمیزی میکروب ها(رنگ آمیزی ساده,افتراقی,مخصوص)
انواع رنگ آمیزی میکروب ها:
یکی از مشخصات مهم باکتری ها که به شناسایی آنها کمک می کند رنگ پذیری آنها با رنگ های مختلفی است که برای این منظور به کار می روند.این رنگ ها یا رنگ های عمومی هستند یعنی همه ی باکتری ها را یکسان رنگ می کنند و یا رنگ های اختصاصی که فقط یک گروه از باکتری ها را به رنگ خاص درمی آورد.
مورفولوژی باکتری ها را می توان به دو صورت مورد بررسی قرار داد یکی به صورت زنده که یا از رنگ استفاد نمی شود و یا از رنگ های حیاتی مثل قرمز خنثی برای رنگ آمیزی باکتری ها استفاده می شود و دیگری به صورت مرده یا به عبارتی فیکس شده که از رنگ ها و روش های مختلفی برای رنگ آمیزی گسترش باکتری استفاده می شود.
تقسیم بندی رنگ ها:
رنگ ها از نظر نوع ترکیب مواد رنگی به دو گروه رنگ های ساده و رنگ های مرکب تقسیم می شوند.
الف)رنگ های ساده:این دسته از رنگها از یک ماده ساخته شده اند
ب)رنگ های مرکب:این گروه از رنگ ها از چند دسته مواد تشکیل شده اند
در تقسیم بندی دیگر رنگ ها را بر اساس میل ترکیبی آنها با اجزا یاخته تقسیم بندی می کنند. در رابطه با این امر رنگ ها از نظر گرایش به اجزا یاخته یا رنگ دوستی هسته و سیتوپلاسم به سه دسته تقسیم می شوند:
۱- رنگ های بازی یا هسته ای : این رنگ ها اصولا یا DNA و یا RNA یاخته ترکیب می شوند.از انواع رنگ های بازی می توان کریستال ویوله , فوشین ,آبی متیلن و سبز متیل را نام برد.کریستال ویوله که بار الکتریکی مثبت دارد به وسیله باکتری دارای بار الکتریکی منفی جذب می شود و آن را رنگ می کند.
۲- رنگ های اسیدی :این رنگ ها شامل سافرانین و فوشین اسیدی و قرمز کنگو هستند و ترکیبات بازی یاخته و اصولاٌ پروتئین های بازی را رنگ می کنند.
۳- رنگ های خنثی : سودان سیاه رنگی است که در چربی محلول است و غالباٌ برای مشخص کردن قطرات چربی و موقعیت آنها در باکتری ها به کار می رود.
در میکروب شناسی غالباٌ از سه نوع رنگ آمیزی استفاده می شود.
الف) رنگ آمیزی ساده
ب) رنگ آمیزی افتراقی
ج) رنگ آمیزی مخصوص
الف) رنگ آمیزی ساده :
در این نوع رنگ آمیزی از یک نوع رنگ استفاده می شود و از این روش می توان برای رنگ کردن کامل سلول یا ساختارهای ویژه آن استفاده کرد. آبی متیلن غالباٌ برای رنگ آمیزی ساده باکتری های فیکس شده به کار می رود و آنها را به رنگ آبی در می آورد.از رنگ های دیگری چون کریستال ویوله و کربول فوشین نیز می توان برای رنگ آمیزی ساده استفاده نمود.گاهی یک رنگ ساده به عنوان رنگ منفی به کار می رود که در این ارتباط می توان به رنگ آمیزی منفی در رابطه با تشخیص کپسول اشاره نمود که زمینه رنگ می گیرد در حالی که کپسول بدون رنگ مشخص می شود.رنگ آمیزی ساده به دو روش می تواند انجام بگیرد:
الف-۱) رنگ آمیزی ساده به روش مثبت (آبی متیلن یا کریستال ویوله )
۱- ابتدا توسط یک لوپ پلاتینی از کلنی یا سوسپانسیون باکتری های موجود یک گسترش بیضی شکل بر روی یک لام تمیز و غیر چرب تهیه نمایید و سپس تا گسترش مذکور در مجاورت هوا خشک شود. گسترش خشک شده را با عبور دادن لام طی ۲ تا ۳ بار از میان شعله فیکس نمایید.هدف از فیکس کردن گسترش چسباندن محکم باکتری های موجود در گسترش به سطح لام می باشد. که این امر به واسطه انعقاد قسمتی از پروتئین های باکتریایی فراهم می گردد. این امر از شسته شدن گسترش در حین رنگ آمیزی جلوگیری می کند.
۲- چند قطره از محلول رنگی آبی متیلن را روی گسترش میکروبی ریخته و پس از دو دقیقه آن را با آب مقطر بشویید.
۳- اجازه دهید تا لام در مجاورت هوا خشک شود.پس از آن در زیر میکروسکوپ به مطالعه ی آن بپردازید.
الف-۲) رنگ آمیزی ساده به روش منفی (مرکب چین یا نگروزین )
همانطوری که قبلاٌ اشاره شد غالباٌ از این روش جهت تشخیص کپسول استفاده می شود که روش آن به ترتیب زیر است:
۱- ابتدا مقداری از نمونه باکتریایی را در یک لوله آزمایش یا حجمی برابر با مرکب چین یا نگروزین مخلوط نمایید.
۲- توسط لوپ مقداری از این سوسپانسیون را بر روی لام گذارده و یک گسترش از آن تهیه نمایید.
۳- اجازه دهید تا گسترش خشک شود سپس توسط میکروسکوپ به مطالعه آن بپردازید در این رنگ آمیزی کپسول با کتری به صورت یک هاله بی رنگ در زمینه تیره مشخص می شود
ب) رنگ آمیزی افتراقی:
رنگ آمیزی است که باکتری های مختلف در برابر آن واکنش مختلفی نشان می دهند و از این طریق می توان آنها را طبقه بندی و مورد شناسایی قرار داد . بهترین روش رنگ آمیزی افتراقی در باکتری شناسی رنگ آمیزی گرم می باشد.
این رتگ آمیزی در سال ۱۸۸۴ توسط پزشک دانمارکی کریستین گرم ابداع گردید. در این رنگ آمیزی باکتری ها به دو گروه تقسیم می شوند: باکتری های گرم مثبت که به رنگ بنش ظاهر می شوند و باکتری های گرم منفی که به رنگ صورتی نمایان می شوند.
در این رنگ آمیزی کریستال ویوله به عنوان رنگ اولیه عمل می کند نمایان می شوند.
در این رنگ آمیزی کریستال ویوله به عنوان رنگ اولیه عمل می کند و به دیواره سلولی باکتری متصل می شود.این اتصال به واسطه کمپلکسی که با (ید) لوگل ایجاد می کند تثبیت می گردد, در واقع ید به عنوان یک موردانت عمل می کند چرا که میل ترکیبی یا جاذبه بین سلول و رنگ را افزایش داده و به نحوی به تثبیت رنگ روی سلول کمک می کند.برخی از گونه های باکتریایی این توانایی را دارند که حتی پس از اضافه کردن رنگ بر ( الکل یا استن ) رنگ کریستال ویوله را حفظ می کنند لذا در رنگ آمیزی گرم به رنگ بنفش در می آیند. این امر به آن علت است که در دیواره باکتری های گرم مثبت ضخیم بودن لایه پپتیدوگلیکان و کم بودن مقدار چربی مانع از نفوذ الکل به دیواره و در عین حال مانع از خروج رسوب رنگی از دیواره می گردد. این در حالی است که دیواره ی سلولی باکتری های گرم منفی به علت وجود مقادیر زیاد چربی و نازک تر بودن لایه پپتیدوگلیکان پس از اضافه نمونه رنگ بر رنگ کریستال ویوله را از دست می دهد و در این موارد بعد از اضافه کردن سافرانین در مرحله آخر به رنگ صورتی در می آیند
ب – ۱) رنگ آمیزی گرم به روش هوکر(Hucker )
1- ابتدا از باکتری مورد نظر یک گسترش نازک تهیه کرده و در هوای آزمایشگاه اجازه دهید تا خشک شود.
۲- گسترش مذکور را فیکس کنید
۳- کریستال ویوله را به مدت ۱ دقیقه روی لام قرار دهید
۴- لام را با آب مقطر شستشو دهید
۵- محلول لوگل را به مدت ۱ دقیقه روی لام اثر دهید
۶- لام را با آب شستشو دهید
۷- از محلول ۲۵ درصد سافرانین به مدت ۲۰ ثانیه بر روی لام استفاده کنید
۸- لام را با آب مقطر شسته و پس از خشک شدن آن در زیرر میکروسکوپ به مطالعه آن بپردازید
باید دانست که شرایط محیط (مانند PH ,مواد غذایی ,سن باکتری) در رنگ پذیری باکتری ها موثرند.برخی از باکتری های گرم مثبت پس از چند روز رشد در محیط غذایی ممکن است در رنگ آمیزی گرم به صورت گرم منفی درآیند همچنین اگر مدت زمان استفاده از رنگ بر زیاد شود ممکن است باکتری های گرم مثبت رنگ اولیه خود را از دست بدهند و به رنگ صورتی درآیند.
ج)رنگ آمیزی مخصوص
از رنگ آمیزی مخصوص جهت مشاهده و بررسی برخی باکتری های خاص و اجزاء داخلی و جانبی آنها استفاده می شود.در این بخش به رنگ آمیزی های خاصی چون زیل نلسون ,لیفسون,شیفر فولتون و آلبرت می توان اشاره نمود
(ج – ۱ ) رنگ آمیزی زیل نلسون
برخی از باکتری ها (باسیل سل ,,جذام و برخی از جنس های نوکاردیا )به واسطه دارا بودن میزان بالایی از چربی به آسانی به روش های معمولی رنگ آمیزی نمی شوند بلکه برای این امر به یک رنگ و حرارت قوی و زمان بیشتری جهت نفوذ رنگ به داخل پیکرشان نیاز دارند.به واسطه این نوع رنگ آمیزی این باکتری ها حتی توسط اسیدهای معدنی و یا اسید الکل رنگ زدایی نمی شوند که به همین جهت واژه اسید فست به آنها اطلاق می گردد.این خاصیت اجاززه می دهد که باکتری ها حتی به تعداد کم در گسترش رنگ شده قابل تشخیص باشند.ترتیب رنگ آمیزی زیل نلسون به قرار زیر است:
۱- یک گسترش ضخیم و یکنواخت از نمونه تهیه و آن را در هوا خشک کرده و سپس توسط شعله فیکس نمایید.
۲- یک کاغذ صافی به ابعاد ۲*۴ سانتی متر یا کمی کوچکتر از لام فراهم کنید.این کاغذ بایستی اسمیر (گسترش ) را بپوشاند تا از رسوب رنگ روی جلوگیری کند.
۳- تمام سطح گسترش را با کربول فوشین (رنگ بازی ) زیل نلسون بپوشانید و به آرامی از سطح زیرین لام توسط شعله حرارت دهید.در زمان حرارت دادن لام بایستی میزان حرارت آنقدر باشد که سطح لام فقط بخار بلند شود و نبایستی رنگ روی لام بجوشد و یا خشک شود در صورتی که به علت حرارت و بخار شدن رنگ میزان رنگ کاهش یابد می توان مجددا روی لام رنگ اضافه کرد
۴- زمان تأثیر رنگ روی گسترش ۵ دقیقه است بعد از آن کاغذ را برداشته و لام را با آب بشویید
۵- توسط اسید الکل رنگ زدایی کنید یا از محلول مزبور آنقدر روی لام قطره قطره می ریزند تا آخرین قطره خارج شده از روی لام شفاف باشد.
۶- از رنگ آبی متیلن به مدت ۳۰ تا ۶۰ ثانیه روی گسترش بریزید
۷- لام را شسته در هوا خشک کنید و با عدسی روغنی به مطالعه پردازید
در این رنگ آمیزی باکتری سل به صورت باسیل ظریف و قرمز رنگی در زمینه آبی مشخص می شود
(ج – ۲) رنگ آمیزی تاژک به روش لیفسون
تاژک ها ضمائمی هستند بسیار شکننده که نسبت به حرارت حساس می باشند و جهت رنگ آمیزی باید با دقت فراوان با انها کار کرد و نیاز به روش های مخصوص می باشد . تاژک ها با میکروسکوپ های معمولی قابل مشاهده نیستند و به این منظور باید قطر انها افزایش یابد تا قابل رویت گردند.این عمل با استفاده از پوشاندن سطح آنها توسط یک ماده موردانت انجام می شود.این ماده اجازه می دهد تا مواد رنگی به خوبی روی آن تثبیت شده و ساختمان نخی شکل تازک بعد از رنگ آمیزی قابل مشاهده گردد.
۱- برای رنگ آمیزی تاژک معمولا کشت ۱۸ الی ۲۴ ساعته باکتری بر روی محیط آگاردار مناسب می باشد که پس از مدت مذکور بر سطح محیط کشت ۲ تا ۳ میلی لیتر آب مقطر سترون ریخته و محلول را در لوله سترون بریزید و ان را به مدت ۱۵ دقیقه در دمای ۳۷ درجه قرار دهید و سپس از قسمت سطحی این سوسپانسیون برداشت کنید
۲- رنگ تازه تهیه شده لیفسون را اختیار کنید
۳- برای رنگ آمیزی روی لام مورد نظر یک مستطیل با ماژیک رسم کنید به طوری که ۳/۲ لام را شامل شود
۴- ۱/۰ میلی لیتر از محلول باکتری را از سطح رویی سوسپانسیون میکروبی برداشته و در یک گوشه مستطیل روی لام قرار دهید
۵- لام را با زاویه ۳۰ تا ۴۰ درجه کج کنید تا محلول از یک طرف لام به سمت دیگر برود لام را به حالت کج در دمای آزمایشگاه خشک کنید. این لام پس از خشک شدن نیازی به ثابت کردن ندارد
۶- قسمت مستطیلی لام را کاملا با رنگ لیفسون بپوشانید و حدود۱۰ تا ۱۵ دقیقه صبر کنید
۷- لام را با آب بشویید برای شستشو بهتر است آب را با پیپت به آرامی روی گسترش بریزید
۸- لام را در دمای آزمایشگاه خشک کنید و آن را توسط عدسی روغنی مورد مطالعه قرار دهید در این حالت تاژک به رنگ صورتی کم رنگ و جسم باکتری پررنگ تر دیده می شود
یاد آوری : از نمونه موارد آزمایش می توانید قطره معلق نیز تهیه کنید تا از متحرک بودن کشت مطمئن شوید
(ج – ۳)رنگ آمیزی گرانول های ولوتین (دانه های متاکروماتیک ) به رو آلبرت
از این رنگ آمیزی جهت تشخیص باسیل کورینه باکتریوم (عامل بیماری دیفتری ) و دیگر اعضا کورینه باکتریوم ها استفاده می شود چرا که این باکتری ها در دیواره خود دارای دانه های متاکروماتیک هستند.ترتیب این رنگ آمیزی به شرح زیر است:
۱- یک گسترش از باکتری مذکور تهیه و پس از فیکس نمودن آن از محلول A به مدت ۵ دقیقه بر روی آن اثر دهید
۲- رنگ را از روی لام بدون شستشوی آن با آب دور بریزید
۳- محلول B را روی لام به مدت ۱ دقیقه اثر دهید
۴- لام را با آب شسته در هوا خشک کرده و توسط عدسی روغنی به مطالعه بپردازید
در این آزمایش گرانول های متاکروماتیک در دو سر باکتری به رنگ سبز تیره تا سیاه و پیکر باکتری به رنگ سبز روشن مشاهده می گردند
محلول A شامل :
آبی تولوئیدین ۱۵ صدم گرم,سبز مالاشیت ۲ صدم گرم,الکل اتیلیک ۹۵ درصد ۲ میلی لیتر ,آب مقطر ۱۰۰ میلی لیتر
محلول B شامل :
یدور پتاسیم ۳ گرم ,ید ۲ گرم ,آب مقطر ۱۰۰ میلی لیتر
 زی درباکتری شناسی است که اولین بار توسط کریسین گرم ابداع شد . دراین رنگ آمیزی باکتریها بر مبنای رنگ باکتری پس ازرنگ آمیزی به دودسته گرم مثبت وگرم منفی تقسیم می شوند . رنگ باکتری پس ازرنگ آمیزی به توانایی حفظ رنگ اول وبه عبارتیبه ساختمان دیواره سلولی باکتری بستگی دارد . دررنگ آمیزی گرم باکتریهای گرم مثبت پس ازرنگ آمیزی به رنگ بنفش وباکتری های گرم منفی به رنگ قرمز مشاهده می شود
زی درباکتری شناسی است که اولین بار توسط کریسین گرم ابداع شد . دراین رنگ آمیزی باکتریها بر مبنای رنگ باکتری پس ازرنگ آمیزی به دودسته گرم مثبت وگرم منفی تقسیم می شوند . رنگ باکتری پس ازرنگ آمیزی به توانایی حفظ رنگ اول وبه عبارتیبه ساختمان دیواره سلولی باکتری بستگی دارد . دررنگ آمیزی گرم باکتریهای گرم مثبت پس ازرنگ آمیزی به رنگ بنفش وباکتری های گرم منفی به رنگ قرمز مشاهده می شود




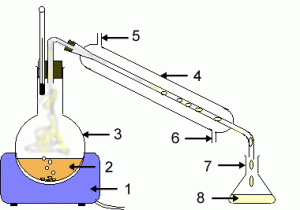
 تئوری آزمایش
تئوری آزمایش و اتر
و اتر  با هم ایزومر گروه عاملی هستند. در ترکیبهایی دیده میشود که یک یا چند لیگاند دارای دو اتم کوئوردیناسیون دهنده باشند، برای مثال یون نیتریک NO-2 میتواند از طریق یک اتم اکسیژن یا از طریق اتم نیتروژن کوئوردیناسیون بدهد. زرد NH3)5CO(NO2)) Cl2)) و قرمز NH3)5-CO-ONO) Cl2))
با هم ایزومر گروه عاملی هستند. در ترکیبهایی دیده میشود که یک یا چند لیگاند دارای دو اتم کوئوردیناسیون دهنده باشند، برای مثال یون نیتریک NO-2 میتواند از طریق یک اتم اکسیژن یا از طریق اتم نیتروژن کوئوردیناسیون بدهد. زرد NH3)5CO(NO2)) Cl2)) و قرمز NH3)5-CO-ONO) Cl2))


 وبسترا و گزلتن که از کربن حاصل گروه OH می باشد.یک الکل نوع اول دارای ۲ هیدروژن α می باشد و می تواند یکی از آنها را از دست داده و به یک آلدئید تبدیل شود و یا هر دو هیدروژن را از دست داده و به یک کربوکسیلیک اسید تبدیل شود.
وبسترا و گزلتن که از کربن حاصل گروه OH می باشد.یک الکل نوع اول دارای ۲ هیدروژن α می باشد و می تواند یکی از آنها را از دست داده و به یک آلدئید تبدیل شود و یا هر دو هیدروژن را از دست داده و به یک کربوکسیلیک اسید تبدیل شود.


 ن دو قسمت آلی رنگ به عنوان پل عمل میکنند و حداقل یکی از این گروهها آروماتیک هستند. توسط گروه کرموفوری (رنگزای) آزو ، میتوان طیف وسیعی از رنگها مثل زرد ، قرمز ، نارنجی ، آبی ، سبز ، بنفش و سیاه را سنتز کرد.
ن دو قسمت آلی رنگ به عنوان پل عمل میکنند و حداقل یکی از این گروهها آروماتیک هستند. توسط گروه کرموفوری (رنگزای) آزو ، میتوان طیف وسیعی از رنگها مثل زرد ، قرمز ، نارنجی ، آبی ، سبز ، بنفش و سیاه را سنتز کرد.